profiletools package¶
Submodules¶
profiletools.CMod module¶
Provides classes for working with Alcator C-Mod data via MDSplus.
-
class
profiletools.CMod.BivariatePlasmaProfile(X_dim=1, X_units=None, y_units='', X_labels=None, y_label='', weightable=True)[source]¶ Bases:
profiletools.core.ProfileClass to represent bivariate (y=f(t, psi)) plasma data.
The first column of X is always time. If the abscissa is ‘RZ’, then the second column is R and the third is Z. Otherwise the second column is the desired abscissa (psinorm, etc.).
-
remake_efit_tree()[source]¶ Remake the EFIT tree.
This is needed since EFIT tree instances aren’t pickleable yet, so to store a
BivariatePlasmaProfilein a pickle file, you must delete the EFIT tree.
-
convert_abscissa(new_abscissa, drop_nan=True, ddof=1)[source]¶ Convert the internal representation of the abscissa to new coordinates.
The target abcissae are what are supported by rho2rho from the eqtools package. Namely,
psinorm Normalized poloidal flux phinorm Normalized toroidal flux volnorm Normalized volume Rmid Midplane major radius r/a Normalized minor radius Additionally, each valid option may be prepended with ‘sqrt’ to return the square root of the desired normalized unit.
Parameters: new_abscissa : str
The new abscissa to convert to. Valid options are defined above.
drop_nan : bool, optional
Set this to True to drop any elements whose value is NaN following the conversion. Default is True (drop NaN elements).
ddof : int, optional
Degree of freedom correction to use when time-averaging a conversion.
-
time_average(**kwargs)[source]¶ Compute the time average of the quantity.
Stores the original bounds of t to self.t_min and self.t_max.
All parameters are passed to
average_data().
-
drop_axis(axis)[source]¶ Drops a selected axis from X.
Parameters: axis : int
The index of the axis to drop.
-
keep_times(times, **kwargs)[source]¶ Keeps only the nearest points to vals along the time axis for each channel.
Parameters: times : array of float
The values the time should be close to.
**kwargs : optional kwargs
All additional kwargs are passed to
keep_slices().
-
add_profile(other)[source]¶ Absorbs the data from another profile object.
Parameters: other :
ProfileProfileto absorb.
-
remove_edge_points(allow_conversion=True)[source]¶ Removes points that are outside the LCFS.
Must be called when the abscissa is a normalized coordinate. Assumes that the last column of self.X is space: so it will do the wrong thing if you have already taken an average along space.
Parameters: allow_conversion : bool, optional
If True and self.abscissa is ‘RZ’, then the profile will be converted to psinorm and the points will be dropped. Default is True (allow conversion).
-
constrain_slope_on_axis(err=0, times=None)[source]¶ Constrains the slope at the magnetic axis of this Profile’s Gaussian process to be zero.
Note that this is accomplished approximately for bivariate data by specifying the slope to be zero at the magnetic axis for a number of points in time.
It is assumed that the Gaussian process has already been created with a call to
create_gp().It is required that the abscissa be either Rmid or one of the normalized coordinates.
Parameters: err : float, optional
The uncertainty to place on the slope constraint. The default is 0 (slope constraint is exact). This could also potentially be an array for bivariate data where you wish to have the uncertainty vary in time.
times : array-like, optional
The times to impose the constraint at. Default is to use the unique time values in X[:, 0].
-
constrain_at_limiter(err_y=0.01, err_dy=0.1, times=None, n_pts=4, expansion=1.25)[source]¶ Constrains the slope and value of this Profile’s Gaussian process to be zero at the GH limiter.
The specific value of X coordinate to impose this constraint at is determined by finding the point of the GH limiter which has the smallest mapped coordinate.
If the limiter location is not found in the tree, the system will instead use R=0.91m, Z=0.0m as the limiter location. This is a bit outside of where the limiter is, but will act as a conservative approximation for cases where the exact position is not available.
Note that this is accomplished approximately for bivariate data by specifying the slope and value to be zero at the limiter for a number of points in time.
It is assumed that the Gaussian process has already been created with a call to
create_gp().The abscissa cannot be ‘Z’ or ‘RZ’.
Parameters: err_y : float, optional
The uncertainty to place on the value constraint. The default is 0.01. This could also potentially be an array for bivariate data where you wish to have the uncertainty vary in time.
err_dy : float, optional
The uncertainty to place on the slope constraint. The default is 0.1. This could also potentially be an array for bivariate data where you wish to have the uncertainty vary in time.
times : array-like, optional
The times to impose the constraint at. Default is to use the unique time values in X[:, 0].
n_pts : int, optional
The number of points outside of the limiter to use. It helps to use three or more points outside the plasma to ensure appropriate behavior. The constraint is applied at n_pts linearly spaced points between the limiter location (computed as discussed above) and the limiter location times expansion. If you set this to one it will only impose the constraint at the limiter. Default is 4.
expansion : float, optional
The factor by which the coordinate of the limiter location is multiplied to get the outer limit of the n_pts constraint points. Default is 1.25.
-
remove_quadrature_points_outside_of_limiter()[source]¶ Remove any of the quadrature points which lie outside of the limiter.
This is accomplished by setting their weights to zero. When
create_gp()is called, it will callGaussianProcess.condense_duplicates()which will remove any points for which all of the weights are zero.This only affects the transformed quantities in self.transformed.
-
get_limiter_locations()[source]¶ Retrieve the location of the GH limiter from the tree.
If the data are not there (they are missing for some old shots), use R=0.91m, Z=0.0m.
-
create_gp(constrain_slope_on_axis=True, constrain_at_limiter=True, axis_constraint_kwargs={}, limiter_constraint_kwargs={}, **kwargs)[source]¶ Create a Gaussian process to handle the data.
Calls
create_gp(), then imposes constraints as requested.Defaults to using a squared exponential kernel in two dimensions or a Gibbs kernel with tanh warping in one dimension.
Parameters: constrain_slope_on_axis : bool, optional
If True, a zero slope constraint at the magnetic axis will be imposed after creating the gp. Default is True (constrain slope).
constrain_at_limiter : bool, optional
If True, a zero slope and value constraint at the GH limiter will be imposed after creating the gp. Default is True (constrain at axis).
axis_constraint_kwargs : dict, optional
The contents of this dictionary are passed as kwargs to
constrain_slope_on_axis().limiter_constraint_kwargs : dict, optional
The contents of this dictionary are passed as kwargs to
constrain_at_limiter().**kwargs : optional kwargs
All remaining kwargs are passed to
Profile.create_gp().
-
compute_a_over_L(X, force_update=False, plot=False, gp_kwargs={}, MAP_kwargs={}, plot_kwargs={}, return_prediction=False, special_vals=0, special_X_vals=0, compute_2=False, **predict_kwargs)[source]¶ Compute the normalized inverse gradient scale length.
Only works on data that have already been time-averaged at the moment.
Parameters: X : array-like
The points to evaluate a/L at.
force_update : bool, optional
If True, a new Gaussian process will be created even if one already exists. Set this if you have added data or constraints since you created the Gaussian process. Default is False (use current Gaussian process if it exists).
plot : bool, optional
If True, a plot of a/L is produced. Default is False (no plot).
gp_kwargs : dict, optional
The entries of this dictionary are passed as kwargs to
create_gp()if it gets called. Default is {}.MAP_kwargs : dict, optional
The entries of this dictionary are passed as kwargs to
find_gp_MAP_estimate()if it gets called. Default is {}.plot_kwargs : dict, optional
The entries of this dictionary are passed as kwargs to plot when plotting the mean of a/L. Default is {}.
return_prediction : bool, optional
If True, the full prediction of the value and gradient are returned in a dictionary. Default is False (just return value and stddev of a/L).
special_vals : int, optional
The number of special return values incorporated into output_transform that should be dropped before computing a/L. This is used so that things like volume averages can be efficiently computed at the same time as a/L. Default is 0 (no extra values).
special_X_vals : int, optional
The number of special points included in the abscissa that should not be included in the evaluation of a/L. Default is 0 (no extra values).
compute_2 : bool, optional
If True, the second derivative and some derived quantities will be computed and added to the output structure (if return_prediction is True). You should almost always have r/a for your abscissa when using this: the expressions for other coordinate systems are not as well-vetted. Default is False (don’t compute second derivative).
**predict_kwargs : optional parameters
All other parameters are passed to the Gaussian process’
predict()method.
-
compute_volume_average(return_std=True, grid=None, npts=400, force_update=False, gp_kwargs={}, MAP_kwargs={}, **predict_kwargs)[source]¶ Compute the volume average of the profile.
Right now only supports data that have already been time-averaged.
Parameters: return_std : bool, optional
If True, the standard deviation of the volume average is computed and returned. Default is True (return mean and stddev of volume average).
grid : array-like, optional
The quadrature points to use when finding the volume average. If these are not provided, a uniform grid over volnorm will be used. Default is None (use uniform volnorm grid).
npts : int, optional
The number of uniformly-spaced volnorm points to use if grid is not specified. Default is 400.
force_update : bool, optional
If True, a new Gaussian process will be created even if one already exists. Set this if you have added data or constraints since you created the Gaussian process. Default is False (use current Gaussian process if it exists).
gp_kwargs : dict, optional
The entries of this dictionary are passed as kwargs to
create_gp()if it gets called. Default is {}.MAP_kwargs : dict, optional
The entries of this dictionary are passed as kwargs to
find_gp_MAP_estimate()if it gets called. Default is {}.**predict_kwargs : optional parameters
All other parameters are passed to the Gaussian process’
predict()method.Returns: mean : float
The mean of the volume average.
std : float
The standard deviation of the volume average. Only returned if return_std is True. Note that this is only sufficient as an error estimate if you separately verify that the integration error is less than this!
-
compute_peaking(return_std=True, grid=None, npts=400, force_update=False, gp_kwargs={}, MAP_kwargs={}, **predict_kwargs)[source]¶ Compute the peaking of the profile.
Right now only supports data that have already been time-averaged.
Uses the definition from Greenwald, et al. (2007):
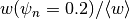 .
.Parameters: return_std : bool, optional
If True, the standard deviation of the volume average is computed and returned. Default is True (return mean and stddev of peaking).
grid : array-like, optional
The quadrature points to use when finding the volume average. If these are not provided, a uniform grid over volnorm will be used. Default is None (use uniform volnorm grid).
npts : int, optional
The number of uniformly-spaced volnorm points to use if grid is not specified. Default is 400.
force_update : bool, optional
If True, a new Gaussian process will be created even if one already exists. Set this if you have added data or constraints since you created the Gaussian process. Default is False (use current Gaussian process if it exists).
gp_kwargs : dict, optional
The entries of this dictionary are passed as kwargs to
create_gp()if it gets called. Default is {}.MAP_kwargs : dict, optional
The entries of this dictionary are passed as kwargs to
find_gp_MAP_estimate()if it gets called. Default is {}.**predict_kwargs : optional parameters
All other parameters are passed to the Gaussian process’
predict()method.
-
-
profiletools.CMod.neCTS(shot, abscissa='RZ', t_min=None, t_max=None, electrons=None, efit_tree=None, remove_edge=False, remove_zeros=True, Z_shift=0.0)[source]¶ Returns a profile representing electron density from the core Thomson scattering system.
Parameters: shot : int
The shot number to load.
abscissa : str, optional
The abscissa to use for the data. The default is ‘RZ’.
t_min : float, optional
The smallest time to include. Default is None (no lower bound).
t_max : float, optional
The largest time to include. Default is None (no upper bound).
electrons : MDSplus.Tree, optional
An MDSplus.Tree object open to the electrons tree of the correct shot. The shot of the given tree is not checked! Default is None (open tree).
efit_tree : eqtools.CModEFITTree, optional
An eqtools.CModEFITTree object open to the correct shot. The shot of the given tree is not checked! Default is None (open tree).
remove_edge : bool, optional
If True, will remove points that are outside the LCFS. It will convert the abscissa to psinorm if necessary. Default is False (keep edge).
remove_zeros: bool, optional
If True, will remove points that are identically zero. Default is True (remove zero points). This was added because clearly bad points aren’t always flagged with a sentinel value of errorbar.
Z_shift: float, optional
The shift to apply to the vertical coordinate, sometimes needed to correct EFIT mapping. Default is 0.0.
-
profiletools.CMod.neETS(shot, abscissa='RZ', t_min=None, t_max=None, electrons=None, efit_tree=None, remove_edge=False, remove_zeros=True, Z_shift=0.0)[source]¶ Returns a profile representing electron density from the edge Thomson scattering system.
Parameters: shot : int
The shot number to load.
abscissa : str, optional
The abscissa to use for the data. The default is ‘RZ’.
t_min : float, optional
The smallest time to include. Default is None (no lower bound).
t_max : float, optional
The largest time to include. Default is None (no upper bound).
electrons : MDSplus.Tree, optional
An MDSplus.Tree object open to the electrons tree of the correct shot. The shot of the given tree is not checked! Default is None (open tree).
efit_tree : eqtools.CModEFITTree, optional
An eqtools.CModEFITTree object open to the correct shot. The shot of the given tree is not checked! Default is None (open tree).
remove_edge : bool, optional
If True, will remove points that are outside the LCFS. It will convert the abscissa to psinorm if necessary. Default is False (keep edge).
remove_zeros: bool, optional
If True, will remove points that are identically zero. Default is True (remove zero points). This was added because clearly bad points aren’t always flagged with a sentinel value of errorbar.
Z_shift: float, optional
The shift to apply to the vertical coordinate, sometimes needed to correct EFIT mapping. Default is 0.0.
-
profiletools.CMod.neTCI(shot, abscissa='r/a', t_min=None, t_max=None, electrons=None, efit_tree=None, quad_points=20, Z_point=-3.0, theta=0.7853981633974483, thin=1, flag_threshold=0.001, ds=0.001)[source]¶ Returns a profile representing electron density from the two color interferometer system.
Parameters: shot : int
The shot number to load.
abscissa : str, optional
The abscissa to use for the data. The default is ‘r/a’.
t_min : float, optional
The smallest time to include. Default is None (no lower bound).
t_max : float, optional
The largest time to include. Default is None (no upper bound).
electrons :
MDSplus.Tree, optionalAn
MDSplus.Treeinstance open to the electrons tree of the correct shot. The shot of the given tree is not checked! Default is None (open tree).efit_tree : :py:class`eqtools.CModEFITTree`, optional
An
eqtools.CModEFITTreeinstance open to the correct shot. The shot of the given tree is not checked! Default is None (open tree).quad_points : int or array of float, optional
The quadrature points to use. If an int, then quad_points linearly- spaced points between 0 and 1.2 will be used. Otherwise, quad_points must be a strictly monotonically increasing array of the quadrature points to use.
Z_point : float
Z coordinate of the starting point of the rays (should be well outside the tokamak). Units are meters.
theta : float
Angle of the chords. Units are radians.
thin : int
Amount by which the data are thinned before computing weights and averages. Default is 1 (no thinning).
flag_threshold : float, optional
The threshold below which points are considered bad. Default is 1e-3.
ds : float, optional
The step size TRIPPy uses to form the beam. Default is 1e-3
-
profiletools.CMod.neTCI_old(shot, abscissa='RZ', t_min=None, t_max=None, electrons=None, efit_tree=None, npts=100, flag_threshold=0.001)[source]¶ Returns a profile representing electron density from the two color interferometer system.
Parameters: shot : int
The shot number to load.
abscissa : str, optional
The abscissa to use for the data. The default is ‘RZ’.
t_min : float, optional
The smallest time to include. Default is None (no lower bound).
t_max : float, optional
The largest time to include. Default is None (no upper bound).
electrons : MDSplus.Tree, optional
An MDSplus.Tree object open to the electrons tree of the correct shot. The shot of the given tree is not checked! Default is None (open tree).
efit_tree : eqtools.CModEFITTree, optional
An eqtools.CModEFITTree object open to the correct shot. The shot of the given tree is not checked! Default is None (open tree).
npts : int, optional
The number of points to use for the line integral. Default is 20.
flag_threshold : float, optional
The threshold below which points are considered bad. Default is 1e-3.
-
profiletools.CMod.neReflect(shot, abscissa='Rmid', t_min=None, t_max=None, electrons=None, efit_tree=None, remove_edge=False, rf=None)[source]¶ Returns a profile representing electron density from the LH/SOL reflectometer system.
Parameters: shot : int
The shot number to load.
abscissa : str, optional
The abscissa to use for the data. The default is ‘Rmid’.
t_min : float, optional
The smallest time to include. Default is None (no lower bound).
t_max : float, optional
The largest time to include. Default is None (no upper bound).
electrons : MDSplus.Tree, optional
An MDSplus.Tree object open to the electrons tree of the correct shot. The shot of the given tree is not checked! Default is None (open tree).
efit_tree : eqtools.CModEFITTree, optional
An eqtools.CModEFITTree object open to the correct shot. The shot of the given tree is not checked! Default is None (open tree).
remove_edge : bool, optional
If True, will remove points that are outside the LCFS. It will convert the abscissa to psinorm if necessary. Default is False (keep edge).
rf : MDSplus.Tree, optional
An MDSplus.Tree object open to the RF tree of the correct shot. The shot of the given tree is not checked! Default is None (open tree).
-
profiletools.CMod.ne(shot, include=['CTS', 'ETS'], TCI_quad_points=None, TCI_flag_threshold=None, TCI_thin=None, TCI_ds=None, **kwargs)[source]¶ Returns a profile representing electron density from both the core and edge Thomson scattering systems.
Parameters: shot : int
The shot number to load.
include : list of str, optional
The data sources to include. Valid options are:
CTS Core Thomson scattering ETS Edge Thomson scattering TCI Two color interferometer reflect SOL reflectometer The default is to include all TS data sources, but not TCI or the reflectometer.
**kwargs
All remaining parameters are passed to the individual loading methods.
-
profiletools.CMod.neTS(shot, **kwargs)[source]¶ Returns a profile representing electron density from both the core and edge Thomson scattering systems.
-
profiletools.CMod.TeCTS(shot, abscissa='RZ', t_min=None, t_max=None, electrons=None, efit_tree=None, remove_edge=False, remove_zeros=True, Z_shift=0.0)[source]¶ Returns a profile representing electron temperature from the core Thomson scattering system.
Parameters: shot : int
The shot number to load.
abscissa : str, optional
The abscissa to use for the data. The default is ‘RZ’.
t_min : float, optional
The smallest time to include. Default is None (no lower bound).
t_max : float, optional
The largest time to include. Default is None (no upper bound).
electrons : MDSplus.Tree, optional
An MDSplus.Tree object open to the electrons tree of the correct shot. The shot of the given tree is not checked! Default is None (open tree).
efit_tree : eqtools.CModEFITTree, optional
An eqtools.CModEFITTree object open to the correct shot. The shot of the given tree is not checked! Default is None (open tree).
remove_edge : bool, optional
If True, will remove points that are outside the LCFS. It will convert the abscissa to psinorm if necessary. Default is False (keep edge).
remove_zeros: bool, optional
If True, will remove points that are identically zero. Default is True (remove zero points). This was added because clearly bad points aren’t always flagged with a sentinel value of errorbar.
Z_shift: float, optional
The shift to apply to the vertical coordinate, sometimes needed to correct EFIT mapping. Default is 0.0.
-
profiletools.CMod.TeETS(shot, abscissa='RZ', t_min=None, t_max=None, electrons=None, efit_tree=None, remove_edge=False, remove_zeros=False, Z_shift=0.0)[source]¶ Returns a profile representing electron temperature from the edge Thomson scattering system.
Parameters: shot : int
The shot number to load.
abscissa : str, optional
The abscissa to use for the data. The default is ‘RZ’.
t_min : float, optional
The smallest time to include. Default is None (no lower bound).
t_max : float, optional
The largest time to include. Default is None (no upper bound).
electrons : MDSplus.Tree, optional
An MDSplus.Tree object open to the electrons tree of the correct shot. The shot of the given tree is not checked! Default is None (open tree).
efit_tree : eqtools.CModEFITTree, optional
An eqtools.CModEFITTree object open to the correct shot. The shot of the given tree is not checked! Default is None (open tree).
remove_edge : bool, optional
If True, will remove points that are outside the LCFS. It will convert the abscissa to psinorm if necessary. Default is False (keep edge).
remove_zeros: bool, optional
If True, will remove points that are identically zero. Default is False (keep zero points). This was added because clearly bad points aren’t always flagged with a sentinel value of errorbar.
Z_shift: float, optional
The shift to apply to the vertical coordinate, sometimes needed to correct EFIT mapping. Default is 0.0.
-
profiletools.CMod.TeFRCECE(shot, rate='s', cutoff=0.15, abscissa='Rmid', t_min=None, t_max=None, electrons=None, efit_tree=None, remove_edge=False)[source]¶ Returns a profile representing electron temperature from the FRCECE system.
Parameters: shot : int
The shot number to load.
rate : {‘s’, ‘f’}, optional
Which timebase to use – the fast or slow data. Default is ‘s’ (slow).
cutoff : float, optional
The cutoff value for eliminating cut-off points. All points with values less than this will be discarded. Default is 0.15.
abscissa : str, optional
The abscissa to use for the data. The default is ‘Rmid’.
t_min : float, optional
The smallest time to include. Default is None (no lower bound).
t_max : float, optional
The largest time to include. Default is None (no upper bound).
electrons : MDSplus.Tree, optional
An MDSplus.Tree object open to the electrons tree of the correct shot. The shot of the given tree is not checked! Default is None (open tree).
efit_tree : eqtools.CModEFITTree, optional
An eqtools.CModEFITTree object open to the correct shot. The shot of the given tree is not checked! Default is None (open tree).
remove_edge : bool, optional
If True, will remove points that are outside the LCFS. It will convert the abscissa to psinorm if necessary. Default is False (keep edge).
-
profiletools.CMod.TeGPC2(shot, abscissa='Rmid', t_min=None, t_max=None, electrons=None, efit_tree=None, remove_edge=False)[source]¶ Returns a profile representing electron temperature from the GPC2 system.
Parameters: shot : int
The shot number to load.
abscissa : str, optional
The abscissa to use for the data. The default is ‘Rmid’.
t_min : float, optional
The smallest time to include. Default is None (no lower bound).
t_max : float, optional
The largest time to include. Default is None (no upper bound).
electrons : MDSplus.Tree, optional
An MDSplus.Tree object open to the electrons tree of the correct shot. The shot of the given tree is not checked! Default is None (open tree).
efit_tree : eqtools.CModEFITTree, optional
An eqtools.CModEFITTree object open to the correct shot. The shot of the given tree is not checked! Default is None (open tree).
remove_edge : bool, optional
If True, will remove points that are outside the LCFS. It will convert the abscissa to psinorm if necessary. Default is False (keep edge).
-
profiletools.CMod.TeGPC(shot, cutoff=0.15, abscissa='Rmid', t_min=None, t_max=None, electrons=None, efit_tree=None, remove_edge=False)[source]¶ Returns a profile representing electron temperature from the GPC system.
Parameters: shot : int
The shot number to load.
cutoff : float, optional
The cutoff value for eliminating cut-off points. All points with values less than this will be discarded. Default is 0.15.
abscissa : str, optional
The abscissa to use for the data. The default is ‘Rmid’.
t_min : float, optional
The smallest time to include. Default is None (no lower bound).
t_max : float, optional
The largest time to include. Default is None (no upper bound).
electrons : MDSplus.Tree, optional
An MDSplus.Tree object open to the electrons tree of the correct shot. The shot of the given tree is not checked! Default is None (open tree).
efit_tree : eqtools.CModEFITTree, optional
An eqtools.CModEFITTree object open to the correct shot. The shot of the given tree is not checked! Default is None (open tree).
remove_edge : bool, optional
If True, will remove points that are outside the LCFS. It will convert the abscissa to psinorm if necessary. Default is False (keep edge).
-
profiletools.CMod.TeMic(shot, cutoff=0.15, abscissa='Rmid', t_min=None, t_max=None, electrons=None, efit_tree=None, remove_edge=False, remove_zeros=True)[source]¶ Returns a profile representing electron temperature from the Michelson interferometer.
Parameters: shot : int
The shot number to load.
abscissa : str, optional
The abscissa to use for the data. The default is ‘Rmid’.
t_min : float, optional
The smallest time to include. Default is None (no lower bound).
t_max : float, optional
The largest time to include. Default is None (no upper bound).
electrons : MDSplus.Tree, optional
An MDSplus.Tree object open to the electrons tree of the correct shot. The shot of the given tree is not checked! Default is None (open tree).
efit_tree : eqtools.CModEFITTree, optional
An eqtools.CModEFITTree object open to the correct shot. The shot of the given tree is not checked! Default is None (open tree).
remove_edge : bool, optional
If True, will remove points that are outside the LCFS. It will convert the abscissa to psinorm if necessary. Default is False (keep edge).
-
profiletools.CMod.Te(shot, include=['CTS', 'ETS', 'FRCECE', 'GPC2', 'GPC', 'Mic'], FRCECE_rate='s', FRCECE_cutoff=0.15, GPC_cutoff=0.15, remove_ECE_edge=True, **kwargs)[source]¶ Returns a profile representing electron temperature from the Thomson scattering and ECE systems.
Parameters: shot : int
The shot number to load.
include : list of str, optional
The data sources to include. Valid options are:
CTS Core Thomson scattering ETS Edge Thomson scattering FRCECE FRC electron cyclotron emission GPC Grating polychromator GPC2 Grating polychromator 2 The default is to include all data sources.
FRCECE_rate : {‘s’, ‘f’}, optional
Which timebase to use for FRCECE – the fast or slow data. Default is ‘s’ (slow).
FRCECE_cutoff : float, optional
The cutoff value for eliminating cut-off points from FRCECE. All points with values less than this will be discarded. Default is 0.15.
GPC_cutoff : float, optional
The cutoff value for eliminating cut-off points from GPC. All points with values less than this will be discarded. Default is 0.15.
remove_ECE_edge : bool, optional
If True, the points outside of the LCFS for the ECE diagnostics will be removed. Note that this overrides remove_edge, if present, in kwargs. Furthermore, this may lead to abscissa being converted to psinorm if an incompatible option was used.
**kwargs
All remaining parameters are passed to the individual loading methods.
-
profiletools.CMod.TeTS(shot, **kwargs)[source]¶ Returns a profile representing electron temperature data from the Thomson scattering system.
Includes both core and edge system.
-
profiletools.CMod.emissAX(shot, system, abscissa='Rmid', t_min=None, t_max=None, tree=None, efit_tree=None, remove_edge=False)[source]¶ Returns a profile representing emissivity from the AXA system.
Parameters: shot : int
The shot number to load.
system : {AXA, AXJ}
The system to use.
abscissa : str, optional
The abscissa to use for the data. The default is ‘Rmid’.
t_min : float, optional
The smallest time to include. Default is None (no lower bound).
t_max : float, optional
The largest time to include. Default is None (no upper bound).
tree : MDSplus.Tree, optional
An MDSplus.Tree object open to the cmod tree of the correct shot. The shot of the given tree is not checked! Default is None (open tree).
efit_tree : eqtools.CModEFITTree, optional
An eqtools.CModEFITTree object open to the correct shot. The shot of the given tree is not checked! Default is None (open tree).
remove_edge : bool, optional
If True, will remove points that are outside the LCFS. It will convert the abscissa to psinorm if necessary. Default is False (keep edge).
-
profiletools.CMod.emiss(shot, include=['AXA', 'AXJ'], **kwargs)[source]¶ Returns a profile representing emissivity.
Parameters: shot : int
The shot number to load.
include : list of str, optional
The data sources to include. Valid options are: {AXA, AXJ}. The default is to include both data sources.
**kwargs
All remaining parameters are passed to the individual loading methods.
-
profiletools.CMod.read_plasma_csv(*args, **kwargs)[source]¶ Returns a profile containing the data from a CSV file.
If your data are bivariate, you must ensure that time ends up being the first column, either by putting it first in your CSV file or by specifying its name first in X_names.
The CSV file can contain metadata lines of the form “name data” or “name data,data,...”. The following metadata are automatically parsed into the correct fields:
shot shot number times comma-separated list of times included in the data t_min minimum time included in the data t_max maximum time included in the data coordinate the abscissa the data are represented as a function of If you don’t provide coordinate in the metadata, the program will try to use the last entry in X_labels to infer the abscissa. If this fails, it will simply set the abscissa to the title of the last entry in X_labels. If you provide your data as a function of R, Z it will look for the last two entries in X_labels to be R and Z once surrounding dollar signs and spaces are removed.
Parameters are the same as
read_csv().
-
profiletools.CMod.read_plasma_NetCDF(*args, **kwargs)[source]¶ Returns a profile containing the data from a NetCDF file.
The file can contain metadata attributes specified in the metadata kwarg. The following metadata are automatically parsed into the correct fields:
shot shot number times comma-separated list of times included in the data t_min minimum time included in the data t_max maximum time included in the data coordinate the abscissa the data are represented as a function of If you don’t provide coordinate in the metadata, the program will try to use the last entry in X_labels to infer the abscissa. If this fails, it will simply set the abscissa to the title of the last entry in X_labels. If you provide your data as a function of R, Z it will look for the last two entries in X_labels to be R and Z once surrounding dollar signs and spaces are removed.
Parameters are the same as
read_NetCDF().
profiletools.core module¶
Provides the base Profile class and other utilities.
-
profiletools.core.average_points(X, y, err_X, err_y, T=None, ddof=1, robust=False, y_method='sample', X_method='sample', weighted=False)[source]¶ Find the average of the points with the given uncertainties using a variety of techniques.
Parameters: X : array, (M, D) or (M, N, D)
Abscissa values to average.
y : array, (M)
Data values to average.
err_X : array, same shape as X
Uncertainty in X.
err_y : array, same shape as y
Uncertainty in y.
T : array, (M, N), optional
Transform for y. Default is None (y is not transformed).
ddof : int, optional
The degree of freedom correction used in computing the standard deviation. The default is 1, the standard Bessel correction to give an unbiased estimate of the variance.
robust : bool, optional
Set this flag to use robust estimators (median, IQR). Default is False.
y_method : {‘sample’, ‘RMS’, ‘total’, ‘of mean’, ‘of mean sample’}, optional
The method to use in computing the uncertainty in the averaged y.
- ‘sample’ computes the sample standard deviation.
- ‘RMS’ computes the root-mean-square of the individual error bars.
- ‘total’ computes the square root of the sum of the sample variance and the mean variance. This is only statistically reasonable if the points represent sample means/variances already.
- ‘of mean’ computes the uncertainty in the mean using error propagation with the given uncertainties.
- ‘of mean sample’ computes the uncertainty in the mean using error propagation with the sample variance. Should not be used with weighted estimators!
Default is ‘sample’ (use sample variance).
X_method : {‘sample’, ‘RMS’, ‘total’, ‘of mean’, ‘of mean sample’}, optional
The method to use in computing the uncertainty in the averaged X. Options are the same as y_method. Default is ‘sample’ (use sample variance).
weighted : bool, optional
Set this flag to use weighted estimators. The weights are 1/err_y^2. Default is False (use unweighted estimators).
Returns: mean_X : array, (D,) or (N, D)
Mean of abscissa values.
mean_y : float
Mean of data values.
err_X : array, same shape as mean_X
Uncertainty in abscissa values.
err_y : float
Uncertainty in data values.
T : array, (N,) or None
Mean of transformation.
-
class
profiletools.core.Channel(X, y, err_X=0, err_y=0, T=None, y_label='', y_units='')[source]¶ Bases:
objectClass to store data from a single channel.
This is particularly useful for storing linearly transformed data, but should work for general data just as well.
Parameters: X : array, (M, N, D)
Abscissa values to use.
y : array, (M,)
Data values.
err_X : array, same shape as X
Uncertainty in X.
err_y : array, (M,)
Uncertainty in data.
T : array, (M, N), optional
Linear transform to get from latent variables to data in y. Default is that y represents untransformed data.
y_label : str, optional
Label for the y data. Default is empty string.
y_units : str, optional
Units of the y data. Default is empty string.
-
keep_slices(axis, vals, tol=None, keep_mixed=False)[source]¶ Only keep the indices closest to given vals.
Parameters: axis : int
The column in X to check values on.
vals : float or 1-d array
The value(s) to keep the points that are nearest to.
keep_mixed : bool, optional
Set this flag to keep transformed quantities that depend on multiple values of X[:, :, axis]. Default is False (drop mixed quantities).
Returns: still_good : bool
Returns True if there are still any points left in the channel, False otherwise.
-
average_data(axis=0, **kwargs)[source]¶ Average the data along the given axis.
Parameters: axis : int, optional
Axis to average along. Default is 0.
**kwargs : optional keyword arguments
All additional kwargs are passed to
average_points().
-
remove_points(conditional)[source]¶ Remove points satisfying conditional.
Parameters: conditional : array, same shape as self.y
Boolean array with True wherever a point should be removed.
Returns: bad_X : array
The removed X values.
bad_err_X : array
The uncertainty in the removed X values.
bad_y : array
The removed y values.
bad_err_y : array
The uncertainty in the removed y values.
bad_T : array
The transformation matrix of the removed y values.
-
-
class
profiletools.core.Profile(X_dim=1, X_units=None, y_units='', X_labels=None, y_label='', weightable=True)[source]¶ Bases:
objectObject to abstractly represent a profile.
Parameters: X_dim : positive int, optional
Number of dimensions of the independent variable. Default value is 1.
X_units : str, list of str or None, optional
Units for each of the independent variables. If X_dim`=1, this should given as a single string, if `X_dim>1, this should be given as a list of strings of length X_dim. Default value is None, meaning a list of empty strings will be used.
y_units : str, optional
Units for the dependent variable. Default is an empty string.
X_labels : str, list of str or None, optional
Descriptive label for each of the independent variables. If X_dim`=1, this should be given as a single string, if `X_dim>1, this should be given as a list of strings of length X_dim. Default value is None, meaning a list of empty strings will be used.
y_label : str, optional
Descriptive label for the dependent variable. Default is an empty string.
weightable : bool, optional
Whether or not it is valid to use weighted estimators on the data, or if the error bars are too suspect for this to be valid. Default is True (allow use of weighted estimators).
Attributes
y ( Array, (M,)) The M dependent variables.X ( Matrix, (M, X_dim)) The M independent variables.err_y ( Array, (M,)) The uncertainty in the M dependent variables.err_X ( Matrix, (M, X_dim)) The uncertainties in each dimension of the M independent variables.channels ( Matrix, (M, X_dim)) The logical groups of points into channels along each of the independent variables.X_dim (positive int) The number of dimensions of the independent variable. X_units (list of str, (X_dim,)) The units for each of the independent variables. y_units (str) The units for the dependent variable. X_labels (list of str, (X_dim,)) Descriptive labels for each of the independent variables. y_label (str) Descriptive label for the dependent variable. weightable (bool) Whether or not weighted estimators can be used. transformed (list of Channel) The transformed quantities associated with theProfileinstance.gp ( gptools.GaussianProcessinstance) The Gaussian process with the local and transformed data included.-
add_data(X, y, err_X=0, err_y=0, channels=None)[source]¶ Add data to the training data set of the
Profileinstance.Will also update the Profile’s Gaussian process instance (if it exists).
Parameters: X : array-like, (M, N)
M independent variables of dimension N.
y : array-like, (M,)
M dependent variables.
err_X : array-like, (M, N), or scalar float, or single array-like (N,), optional
Non-negative values only. Error given as standard deviation for each of the N dimensions in the M independent variables. If a scalar is given, it is used for all of the values. If a single array of length N is given, it is used for each point. The default is to assign zero error to each point.
err_y : array-like (M,) or scalar float, optional
Non-negative values only. Error given as standard deviation in the M dependent variables. If err_y is a scalar, the data set is taken to be homoscedastic (constant error). Otherwise, the length of err_y must equal the length of y. Default value is 0 (noiseless observations).
channels : dict or array-like (M, N)
Keys to logically group points into “channels” along each dimension of X. If not passed, channels are based simply on which points have equal values in X. If only certain dimensions have groupings other than the simple default equality conditions, then you can pass a dict with integer keys in the interval [0, X_dim-1] whose values are the arrays of length M indicating the channels. Otherwise, you can pass in a full (M, N) array.
Raises: ValueError
Bad shapes for any of the inputs, negative values for err_y or n.
-
add_profile(other)[source]¶ Absorbs the data from one profile object.
Parameters: other :
ProfileProfileto absorb.
-
drop_axis(axis)[source]¶ Drops a selected axis from X.
Parameters: axis : int
The index of the axis to drop.
-
keep_slices(axis, vals, tol=None, **kwargs)[source]¶ Keeps only the nearest points to vals along the given axis for each channel.
Parameters: axis : int
The axis to take the slice(s) of.
vals : array of float
The values the axis should be close to.
tol : float or None
Tolerance on nearest values – if the nearest value is farther than this, it is not kept. If None, this is not applied.
**kwargs : optional kwargs
All additional kwargs are passed to
keep_slices().
-
average_data(axis=0, **kwargs)[source]¶ Computes the average of the profile over the desired axis.
If X_dim is already 1, this returns the average of the quantity. Otherwise, the
Profileis mutated to contain the desired averaged data. err_X and err_y are populated with the standard deviations of the respective quantities. The averaging is carried out within the groupings defined by the channels attribute.Parameters: axis : int, optional
The index of the dimension to average over. Default is 0.
**kwargs : optional kwargs
All additional kwargs are passed to
average_points().
-
plot_data(ax=None, label_axes=True, **kwargs)[source]¶ Plot the data stored in this Profile. Only works for X_dim = 1 or 2.
Parameters: ax : axis instance, optional
Axis to plot the result on. If no axis is passed, one is created. If the string ‘gca’ is passed, the current axis (from plt.gca()) is used. If X_dim = 2, the axis must be 3d.
label_axes : bool, optional
If True, the axes will be labelled with strings constructed from the labels and units set when creating the Profile instance. Default is True (label axes).
**kwargs : extra plotting arguments, optional
Extra arguments that are passed to errorbar/errorbar3d.
Returns: The axis instance used.
-
remove_points(conditional)[source]¶ Remove points where conditional is True.
Note that this does NOT remove anything from the GP – you either need to call
create_gp()again or act manually on thegpattribute.Also note that this does not include any provision for removing points that represent linearly-transformed quantities – you will need to operate directly on
transformedto remove such points.Parameters: conditional : array-like of bool, (M,)
Array of booleans corresponding to each entry in y. Where an entry is True, that value will be removed.
Returns: X_bad : matrix
Input values of the bad points.
y_bad : array
Bad values.
err_X_bad : array
Uncertainties on the abcissa of the bad values.
err_y_bad : array
Uncertainties on the bad values.
-
remove_outliers(thresh=3, check_transformed=False, force_update=False, mask_only=False, gp_kwargs={}, MAP_kwargs={}, **predict_kwargs)[source]¶ Remove outliers from the Gaussian process.
The Gaussian process is created if it does not already exist. The chopping of values assumes that any artificial constraints that have been added to the GP are at the END of the GP’s data arrays.
The values removed are returned.
Parameters: thresh : float, optional
The threshold as a multiplier times err_y. Default is 3 (i.e., throw away all 3-sigma points).
check_transformed : bool, optional
Set this flag to check if transformed quantities are outliers. Default is False (don’t check transformed quantities).
force_update : bool, optional
If True, a new Gaussian process will be created even if one already exists. Set this if you have added data or constraints since you created the Gaussian process. Default is False (use current Gaussian process if it exists).
mask_only : bool, optional
Set this flag to return only a mask of the non-transformed points that are flagged. Default is False (completely remove bad points). In either case, the bad transformed points will ALWAYS be removed if check_transformed is True.
gp_kwargs : dict, optional
The entries of this dictionary are passed as kwargs to
create_gp()if it gets called. Default is {}.MAP_kwargs : dict, optional
The entries of this dictionary are passed as kwargs to
find_gp_MAP_estimate()if it gets called. Default is {}.**predict_kwargs : optional parameters
All other parameters are passed to the Gaussian process’
predict()method.Returns: X_bad : matrix
Input values of the bad points.
y_bad : array
Bad values.
err_X_bad : array
Uncertainties on the abcissa of the bad values.
err_y_bad : array
Uncertainties on the bad values.
transformed_bad : array of
ChannelTransformed points that were removed.
-
remove_extreme_changes(thresh=10, logic='and', mask_only=False)[source]¶ Removes points at which there is an extreme change.
Only for univariate data!
This operation is performed by looking for points who differ by more than thresh * err_y from each of their neighbors. This operation will typically only be useful with large values of thresh. This is useful for eliminating bad channels.
Note that this will NOT update the Gaussian process.
Parameters: thresh : float, optional
The threshold as a multiplier times err_y. Default is 10 (i.e., throw away all 10-sigma changes).
logic : {‘and’, ‘or’}, optional
Whether the logical operation performed should be an and or an or when looking at left-hand and right-hand differences. ‘and’ is more conservative, but ‘or’ will help if you have multiple bad channels in a row. Default is ‘and’ (point must have a drastic change in both directions to be rejected).
mask_only : bool, optional
If True, only the boolean mask indicated where the bad points are will be removed, and it is up to the user to remove them. Default is False (actually remove the bad points).
-
create_gp(k=None, noise_k=None, upper_factor=5, lower_factor=5, x0_bounds=None, mask=None, k_kwargs={}, **kwargs)[source]¶ Create a Gaussian process to handle the data.
Parameters: k :
Kernelinstance, optionalCovariance kernel (from
gptools) with the appropriate number of dimensions, or None. If None, a squared exponential kernel is used. Can also be a string from the following table:SE Squared exponential gibbstanh Gibbs kernel with tanh warping RQ Rational quadratic SEsym1d 1d SE with symmetry constraint The bounds for each hyperparameter are selected as follows:
sigma_f [1/lower_factor, upper_factor]*range(y) l1 [1/lower_factor, upper_factor]*range(X[:, 1]) ... And so on for each length scale Here, eps is sys.float_info.epsilon. The initial guesses for each parameter are set to be halfway between the upper and lower bounds. For the Gibbs kernel, the uniform prior for sigma_f is used, but gamma priors are used for the remaining hyperparameters. Default is None (use SE kernel).
noise_k :
Kernelinstance, optionalThe noise covariance kernel. Default is None (use the default zero noise kernel, with all noise being specified by err_y).
upper_factor : float, optional
Factor by which the range of the data is multiplied for the upper bounds on both length scales and signal variances. Default is 5, which seems to work pretty well for C-Mod data.
lower_factor : float, optional
Factor by which the range of the data is divided for the lower bounds on both length scales and signal variances. Default is 5, which seems to work pretty well for C-Mod data.
x0_bounds : 2-tuple, optional
Bounds to use on the x0 (transition location) hyperparameter of the Gibbs covariance function with tanh warping. This is the hyperparameter that tends to need the most tuning on C-Mod data. Default is None (use range of X).
mask : array of bool, optional
Boolean mask of values to actually include in the GP. Default is to include all values.
k_kwargs : dict, optional
All entries are passed as kwargs to the constructor for the kernel if a kernel instance is not provided.
**kwargs : optional kwargs
All additional kwargs are passed to the constructor of
gptools.GaussianProcess.
-
find_gp_MAP_estimate(force_update=False, gp_kwargs={}, **kwargs)[source]¶ Find the MAP estimate for the hyperparameters of the Profile’s Gaussian process.
If this
Profileinstance does not already have a Gaussian process, it will be created. Note that the user is responsible for manually updating the Gaussian process if more data are added or theProfileis otherwise mutated. This can be accomplished directly using the force_update keyword.Parameters: force_update : bool, optional
If True, a new Gaussian process will be created even if one already exists. Set this if you have added data or constraints since you created the Gaussian process. Default is False (use current Gaussian process if it exists).
gp_kwargs : dict, optional
The entries of this dictionary are passed as kwargs to
create_gp()if it gets called. Default is {}.**kwargs : optional parameters
All other parameters are passed to the Gaussian process’
optimize_hyperparameters()method.
-
plot_gp(force_update=False, gp_kwargs={}, MAP_kwargs={}, **kwargs)[source]¶ Plot the current state of the Profile’s Gaussian process.
If this
Profileinstance does not already have a Gaussian process, it will be created. Note that the user is responsible for manually updating the Gaussian process if more data are added or theProfileis otherwise mutated. This can be accomplished directly using the force_update keyword.Parameters: force_update : bool, optional
If True, a new Gaussian process will be created even if one already exists. Set this if you have added data or constraints since you created the Gaussian process. Default is False (use current Gaussian process if it exists).
gp_kwargs : dict, optional
The entries of this dictionary are passed as kwargs to
create_gp()if it gets called. Default is {}.MAP_kwargs : dict, optional
The entries of this dictionary are passed as kwargs to
find_gp_MAP_estimate()if it gets called. Default is {}.**kwargs : optional parameters
All other parameters are passed to the Gaussian process’
plot()method.
-
smooth(X, n=0, force_update=False, plot=False, gp_kwargs={}, MAP_kwargs={}, **kwargs)[source]¶ Evaluate the underlying smooth curve at a given set of points using Gaussian process regression.
If this
Profileinstance does not already have a Gaussian process, it will be created. Note that the user is responsible for manually updating the Gaussian process if more data are added or theProfileis otherwise mutated. This can be accomplished directly using the force_update keyword.Parameters: X : array-like (N, X_dim)
Points to evaluate smooth curve at.
n : non-negative int, optional
The order of derivative to evaluate at. Default is 0 (return value). See the documentation on
gptools.GaussianProcess.predict().force_update : bool, optional
If True, a new Gaussian process will be created even if one already exists. Set this if you have added data or constraints since you created the Gaussian process. Default is False (use current Gaussian process if it exists).
plot : bool, optional
If True,
gptools.GaussianProcess.plot()is called to produce a plot of the smoothed curve. Otherwise,gptools.GaussianProcess.predict()is called directly.gp_kwargs : dict, optional
The entries of this dictionary are passed as kwargs to
create_gp()if it gets called. Default is {}.MAP_kwargs : dict, optional
The entries of this dictionary are passed as kwargs to
find_gp_MAP_estimate()if it gets called. Default is {}.**kwargs : optional parameters
All other parameters are passed to the Gaussian process’
plot()orpredict()method according to the state of the plot keyword.Returns: ax : axis instance
The axis instance used. This is only returned if the plot keyword is True.
mean :
Array, (M,)Predicted GP mean. Only returned if full_output is False.
std :
Array, (M,)Predicted standard deviation, only returned if return_std is True and full_output is False.
full_output : dict
Dictionary with fields for mean, std, cov and possibly random samples. Only returned if full_output is True.
-
-
profiletools.core.read_csv(filename, X_names=None, y_name=None, metadata_lines=None)[source]¶ Reads a CSV file into a
Profile.If names are not provided for the columns holding the X and y values and errors, the names are found automatically by looking at the header row, and are used in the order found, with the last column being y. Otherwise, the columns will be read in the order specified. The column names should be of the form “name [units]”, which will be automatically parsed to populate the
Profile. In either case, there can be a corresponding column “err_name [units]” which holds the 1-sigma uncertainty in that quantity. There can be an arbitrary number of lines of metadata at the beginning of the file which are read into themetadataattribute of theProfilecreated. This is most useful when usingBivariatePlasmaProfileas you can store the shot and time window.Parameters: X_names : list of str, optional
Ordered list of the column names containing the independent variables. The default behavior is to infer the names and ordering from the header of the CSV file. See the discussion above. Note that if you provide X_names you must also provide y_name.
y_name : str, optional
Name of the column containing the dependent variable. The default behavior is to infer this name from the header of the CSV file. See the discussion above. Note that if you provide y_name you must also provide X_names.
metadata_lines : non-negative int, optional
Number of lines of metadata to read from the beginning of the file. These are read into the
metadataattribute of the profile created.
-
profiletools.core.read_NetCDF(filename, X_names, y_name, metadata=[])[source]¶ Reads a NetCDF file into a
Profile.The file must contain arrays of equal length for each of the independent and the dependent variable. The units of each variable can either be specified as the units attribute on the variable, or the variable name can be of the form “name [units]”, which will be automatically parsed to populate the
Profile. For each independent and the dependent variable there can be a corresponding column “err_name” or “err_name [units]” which holds the 1-sigma uncertainty in that quantity. There can be an arbitrary number of metadata attributes in the file which are read into the corresponding attributes of theProfilecreated. This is most useful when usingBivariatePlasmaProfileas you can store the shot and time window. Be careful that you do not overwrite attributes needed by the class, however!Parameters: X_names : list of str
Ordered list of the column names containing the independent variables. See the discussion above regarding name conventions.
y_name : str
Name of the column containing the dependent variable. See the discussion above regarding name conventions.
metadata : list of str, optional
List of attribute names to read into the corresponding attributes of the
Profilecreated.
-
profiletools.core.errorbar3d(ax, x, y, z, xerr=None, yerr=None, zerr=None, **kwargs)[source]¶ Draws errorbar plot of z(x, y) with errorbars on all variables.
Parameters: ax : 3d axis instance
The axis to draw the plot on.
x : array, (M,)
x-values of data.
y : array, (M,)
y-values of data.
z : array, (M,)
z-values of data.
xerr : array, (M,), optional
Errors in x-values. Default value is 0.
yerr : array, (M,), optional
Errors in y-values. Default value is 0.
zerr : array, (M,), optional
Errors in z-values. Default value is 0.
**kwargs : optional
Extra arguments are passed to the plot command used to draw the datapoints.
-
profiletools.core.unique_rows(arr)[source]¶ Returns a copy of arr with duplicate rows removed.
From Stackoverflow “Find unique rows in numpy.array.”
Parameters: arr :
Array, (m, n). The array to find the unique rows of.Returns: unique :
Array, (p, n) where p <= mThe array arr with duplicate rows removed.
-
profiletools.core.get_nearest_idx(v, a)[source]¶ Returns the array of indices of the nearest value in a corresponding to each value in v.
Parameters: v : Array
Input values to match to nearest neighbors in a.
a : Array
Given values to match against.
Returns: Indices in a of the nearest values to each value in v. Has the same shape as v.
-
class
profiletools.core.RejectionFunc(mask, positivity=True, monotonicity=True)[source]¶ Bases:
objectRejection function for use with full_MC mode of
GaussianProcess.predict().Parameters: mask : array of bool
Mask for the values to include in the test.
positivity : bool, optional
Set this to True to impose a positivity constraint on the sample. Default is True.
monotonicity : bool, optional
Set this to True to impose a positivity constraint on the samples. Default is True.
-
profiletools.core.leading_axis_product(w, x)[source]¶ Perform a product along the leading axis, as is needed when applying weights.
-
profiletools.core.meanw(x, weights=None, axis=None)[source]¶ Weighted mean of data.
Defined as
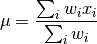
Parameters: x : array-like
The vector to find the mean of.
weights : array-like, optional
The weights. Must be broadcastable with x. Default is to use the unweighted mean.
axis : int, optional
The axis to take the mean along. Default is to use the whole data set.
-
profiletools.core.varw(x, weights=None, axis=None, ddof=1, mean=None)[source]¶ Weighted variance of data.
Defined (for ddof = 1) as
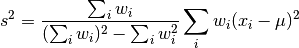
Parameters: x : array-like
The vector to find the mean of.
weights : array-like, optional
The weights. Must be broadcastable with x. Default is to use the unweighted mean.
axis : int, optional
The axis to take the mean along. Default is to use the whole data set.
ddof : int, optional
The degree of freedom correction to use. If no weights are given, this is the standard Bessel correction. If weights are given, this uses an approximate form based on the assumption that the weights are inverse variances for each data point. In this case, the value has no effect other than being True or False. Default is 1 (apply correction assuming normal noise dictated weights).
mean : array-like, optional
The weighted mean to use. If you have already computed the weighted mean with
meanw(), you can pass the result in here to save time.
-
profiletools.core.stdw(*args, **kwargs)[source]¶ Weighted standard deviation of data.
Defined (for ddof = 1) as
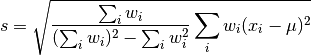
Parameters: x : array-like
The vector to find the mean of.
weights : array-like, optional
The weights. Must be broadcastable with x. Default is to use the unweighted mean.
axis : int, optional
The axis to take the mean along. Default is to use the whole data set.
ddof : int, optional
The degree of freedom correction to use. If no weights are given, this is the standard Bessel correction. If weights are given, this uses an approximate form based on the assumption that the weights are inverse variances for each data point. In this case, the value has no effect other than being True or False. Default is 1 (apply correction assuming normal noise dictated weights).
mean : array-like, optional
The weighted mean to use. If you have already computed the weighted mean with
meanw(), you can pass the result in here to save time.
-
profiletools.core.robust_std(y, axis=None)[source]¶ Computes the robust standard deviation of the given data.
This is defined as
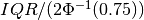 , where
, where 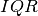 is the
interquartile range and
is the
interquartile range and 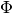 is the inverse CDF of the standard
normal. This is an approximation based on the assumption that the data are
Gaussian, and will have the effect of diminishing the effect of outliers.
is the inverse CDF of the standard
normal. This is an approximation based on the assumption that the data are
Gaussian, and will have the effect of diminishing the effect of outliers.Parameters: y : array-like
The data to find the robust standard deviation of.
axis : int, optional
The axis to find the standard deviation along. Default is None (find from whole data set).
-
profiletools.core.scoreatpercentilew(x, p, weights)[source]¶ Computes the weighted score at the given percentile.
Does not work on small data sets!
Parameters: x : array
Array of data to apply to. Only works properly on 1d data!
p : float or array of float
Percentile(s) to find.
weights : array, same shape as x
The weights to apply to the values in x.
-
profiletools.core.medianw(x, weights=None, axis=None)[source]¶ Computes the weighted median of the given data.
Does not work on small data sets!
Parameters: x : array
Array of data to apply to. Only works properly on 1d, 2d and 3d data.
weights : array, optional
Weights to apply to the values in x. Default is to use an unweighted estimator.
axis : int, optional
The axis to take the median along. Default is None (apply to flattened array).
-
profiletools.core.robust_stdw(x, weights=None, axis=None)[source]¶ Computes the weighted robust standard deviation from the weighted IQR.
Does not work on small data sets!
Parameters: x : array
Array of data to apply to. Only works properly on 1d, 2d and 3d data.
weights : array, optional
Weights to apply to the values in x. Default is to use an unweighted estimator.
axis : int, optional
The axis to take the robust standard deviation along. Default is None (apply to flattened array).